
肌萎缩侧索硬化
Pathogenesis of ALS
肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)是一种罕见的累及上、下运动神经元的神经退行性疾病,临床表现为肌肉进行性无力、萎缩,患者最终因吞咽、呼吸困难而死亡。ALS的病因众多,其中遗传因素相关性极大。神经元中蛋白质内稳态失衡、异常蛋白质的朊病毒样增殖和传播、线粒体功能障碍、谷氨酸介导的兴奋性毒性、神经元内物质运输障碍是目前公认的发病机制。对发病机制相关基因突变的研究将搭起ALS分子水平研究和细胞水平研究的桥梁,从而加深对ALS的发生和发展及基因突变在其中扮演角色的了解,并为疾病的治疗提供新的思路与启示。

血友病
Pathogenesis of hemophilia
血友病乙( Hemophilia B,HB) 为 X 染色体连锁的隐性遗传性出血性疾病,其发病机制多为凝血因子 IX ( F9) 基因突变致凝血因子 IX 缺乏或结构功能的异常。本研究对 40 例明确诊断为血友病乙的男性患者进行基因序列分析以寻找其分子发病机制并为遗传学咨询奠定基础。2009 年 6 月 - 2012 年 6 月收集血友病乙男性患者 40例,均根据临床表现、血浆凝血因子 IX 活性检测及家族史等资料确诊。常规方法收集患者及亲属血样,分离血细胞和血浆,提取 DNA,用聚合酶联反应( PCR) 扩增结合直接测序法分析先证者的 F9 基因的所有外显子及其侧翼序列,并将测序结果和国际 F9 基因突变数据库进行比对,发现突变后排除多态性。结果表明,对 40 例 HB 患者基因分析共发现 34 种基因突变,突变类型包括插入框移突变 2 种,无义突变 6 种,剪切位点的突变 2 种,错义突变 24种,其中 29 种突变为已报道的突变类型,5 种突变在国际上未见报道。34 种突变中发生在信号肽序列的有 2 种,前肽及 γ-羧基化结构域的有 7 种突变,表皮生长因子样结构域有 7 种,活化肽区域有 3 种,丝氨酸蛋白酶催化结构域有 15 种突变。结论: 40 例 HB 患者的基因分析表明 F9 基因突变具有明显的异质性,且错义突变仍是目前 F9 基因的主要突变方式,与重型 HB 有更密切的联系。F9 基因突变所常见的外显子部位与他们编码的氨基酸在蛋白行使功能时所起的作用有关。直接进行基因测序及短串联重复序列连锁分析将会对血友病乙的携带者诊断及产前诊断提供了一种快速、准确的诊断方法。

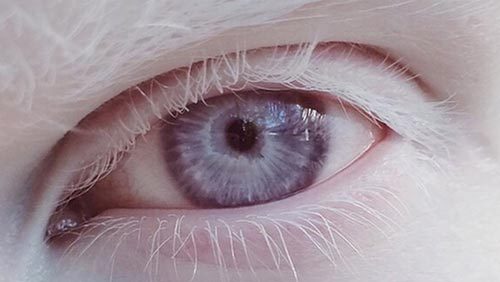
白化病
Pathogenesis of vernacular disease
白化病是具有色素缺失表现的-类遗传性疾病的总称,如伴有其他异常,形成具有白化病表型的综合征,如Hermansky-Pudlak综合征和Chediak -Higashi综合征。H 'rnansky-Pudlak综合征有眼皮肤白化病表现、出血倾向并可伴有其他异常,分为1 ~6个亚型,除Hern hansky-Pudlak综合征2的致病基因功能已知外,其他基因作用尚不清楚。Chediak-Higashi综合征是另-种具有白化病和免疫缺陷等异常的综合征,但目前对其发病机制了解不多。
21-羟化酶缺乏症
Clinical Advances in the Pharmacotherapy of 21- Hydroxylase Deficiency
21-羟化酶缺乏症(21-OHD)是一种罕见的常染色体隐性遗传病,主要由于肾上腺皮质激素不足与雄激素过量所致,治疗上主要采用糖皮质激素及盐皮质激素替代治疗,但氢化可的松的剂量调整及治疗监测对临床医生具有较大挑战,本文主要针对21-羟化酶缺乏症药物治疗进展进行综述。
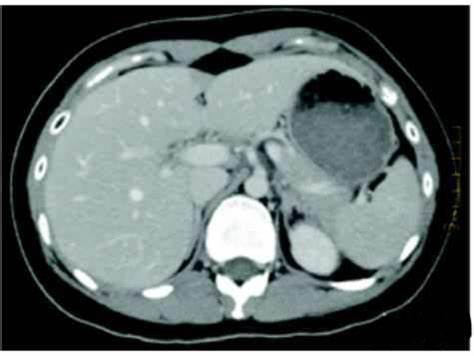
Alport 综合征
Clinical manifestations of Alport syndrome
Alport 综合征是一种遗传性基底膜病变,患者具有 特征性的肾脏、眼部及耳部病变,又称“遗传性肾病”、 “眼耳肾综合征”、“遗传性血尿肾病耳聋综合征”等。 Alport 综合征患者会进展为终末性肾病,在美国儿童终 末期肾病中占 3% ,成人终末期肾病中占 0. 2%
精氨酸酶缺乏症
Arginase deficiency
精氨酸酶缺乏症典型的临床表现为进行性痉挛性瘫痪、认知能力的退化、身材矮小。它与其他类型的尿素循环障碍临床表现不同,高氨血症较为少见或表现往往比较轻微,新生儿期发病的病例少见报道。婴儿很少出现严重的高氨血症或高氨血症昏迷,但也会因高蛋白饮食或感染或禁食等应激状态导致严重的高氨血症,从而出现烦躁不安、嗜睡、拒绝进食、呼吸困难、运动障碍、呕吐甚至昏迷等症状。幼儿出现恶心、呕吐、吞咽困难、动作笨拙、易跌倒等症状。如未经及时诊断和治疗,症状则进行性加重,出现痉挛性瘫痪、昏迷、惊厥、精神和生长发育迟滞等症状。超过一半的患儿

天使综合征
Pathogenesis of Angelman syndrome
天使综合征(angelman syndrome,AS)是一.种较为罕见的严重神经精神系统疾病,多见于儿童,临床上主要表现为发育迟缓、智力低下、孤独内向、吞咽困难、语言和运动功能障碍、癫痫等症状。染色体15q11-q13 区段的UBE3A基因异常是该病发生的主要因素,但是对该病的发病机制尚未形成统一一的学说,并且这种神经精神性疾病的治疗仍然是临床中难点之一,目前已有资料反映出分子靶向治疗具有更好的前景,可能会是未来工作的重点与突破点。本文就天使综合征的发病机制与治疗研究进展进行综述,提取出目前较为有价值的发病机制假说,探讨潜在的治疗方案,并着重关注本病的分子靶向治疗,借以帮助临床医生加深对本病的认识。
